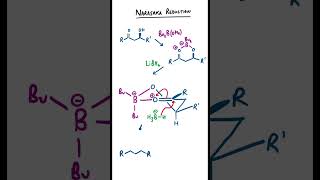
Disconnecting Dactylol - Retrosynthesis

Organic Chemistry: Diastereoselective retrosynthetic analysis of racemic dactylol, a terpenoid natural product.
More retrosynthesis videos available here: • Retrosynthesis
If you enjoyed the video and want to know when new videos drop, please subscribe to my channel here: https://www.youtube.com/CasualChemist...
Reference:
Discussion inspired by this paper:
J. Org. Chem. 1996, 61, 8746
https://doi.org/10.1021/jo961600c
This bicyclic molecule has a tricky 8membered ring to synthesise and also a not particularly favourable 8,5trans ring junction. Mediumsized rings tend to be hard to synthesise as ring closure of a linear precursor is made by the fact that the product has transannular strain that can overturn the good enthalpy change of forming a new sigma bond. Also, the two ends find it hard to meet in the correct orientation for e.g. a substitution reaction as all the degrees of rotational freedom around the other sigma bonds means that there’s a large number of populated conformations that are not reactive for ring closure.
Luckily, we can identify the alkene in the 8membered ring as a key point for disconnection in the retrosynthesis. We can use a ring closing metathesis reaction to cleave through the C=C bond and leave us with two separate chains with new alkenes. Ring closing metathesis (RCM) is good at forming pretty much any ring size by this sort of this disconnection, easily up to 20membered rings, and probably beyond, provided that the reaction conditions are dilute enough to prevent intermolecular reaction (polymerisation) and allow the intramolecular ring closure to occur at an acceptable rate of reaction.
After this ring closing metathesis disconnection, we are left with a tertiary alcohol with three different substituents. These alcohol motifs are easily constructed using Grignard reagents acting as hard nucleophiles on carbonyls, here a ketone. The diastereoselectivity for the bottom face of the cyclopentanone ring will be largely controlled by the adjacent alpha stereocentre on steric grounds. That alkyl group, even though it is likely pseudoequatorial, provides steric hindrance on the top face when compared to the back face which only has a hydrogen at this carbon atom. This leaves us with an substituted cyclopentanone with its groups arranged in an anti fashion. In this retrosynthesis, we are heading to the racemic product, so we just need to make the correct diastereomer next and not a specific enantiomer. As the Grignard addition to the ketone was diastereoselective, if we wanted to make a single enantiomer of the final natural product, we should try to make the next part of the retrosynthetic analysis enantioselective or enantiospecific.
The anti alpha,beta substitution pattern on the cyclopentanone can be disconnected back to the unsaturated cyclopentenone. Addition of a methyl cuprate will add a methyl group to one face (50:50 top face to bottom face) as if a conjugate addition has occurred. This will leave an enolate product with an adjacent new stereocentre right next to the nucleophilic carbon centre. Hence if we don’t do a work up and instead add an electrophile to this enolate intermediate directly, the enolate will react as a nucleophile and pick up a new substituent on the opposite face to the initial methyl group that came from the cuprate organometallic. So we will always form the anti diastereomer from this type of chemistry.
#chemistry #orgo #organicchemistry #ochem #synthesis #retrosynthesis #science #stem #education









![How to Memorize Organic Chemistry Reactions and Reagents [Workshop Recording]](https://i.ytimg.com/vi/030FUb25fSs/mqdefault.jpg)